
TIES is a collection of software packages which can be used to calculate protein ligand binding free energies with physics based alchemical methods. TIES stands for thermodynamic integration with enhanced sampling and this method focuses on the use of ensemble simulations to control for the alleotoric errors in molecular dynamics simulations.
Within the TIES collection there are two packages: TIES 20 and TIES MD. TIES 20 can be used to build all input for TIES MD. TIES MD is an implementation of the TIES protocol that is built on OpenMM and NAMD. Please see the links below for more information on both these programs.
Access to TIES
TIES is free to use and sign up for, by registering using the red TIES 20 button below when using an academic email address. If you’d like to use TIES and you don’t currently have an academic email, please drop us an email on contact@ccs-ties.org. Non-academics currently need to be activated manually. Likewise TIES MD can be accessed using the red TIES MD button and can be used without an account.
News Highlights

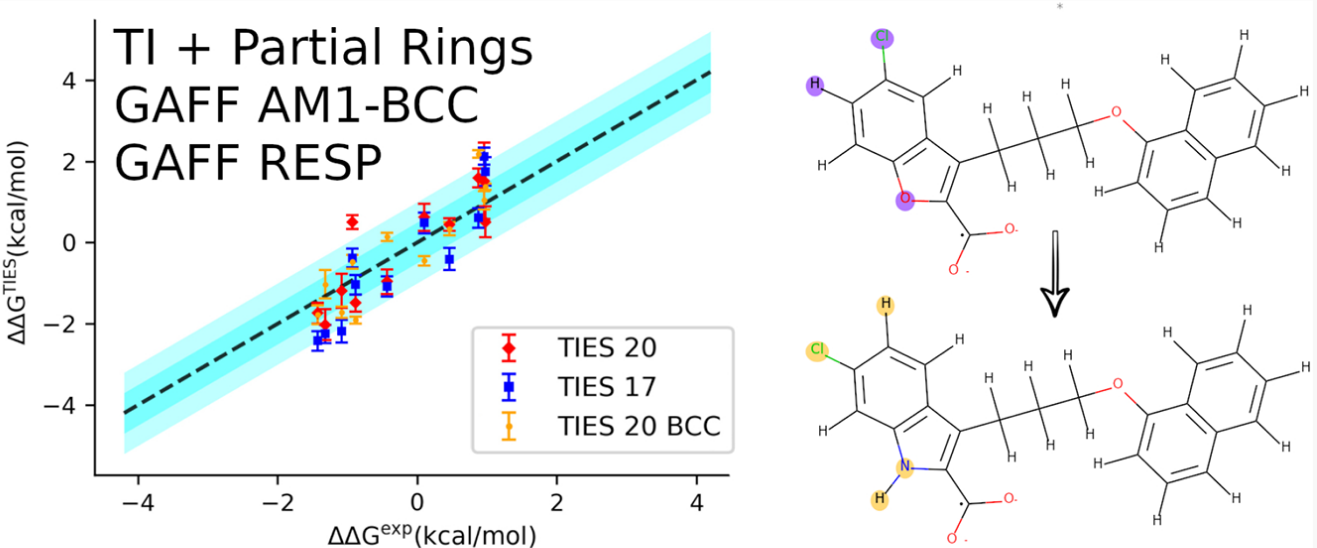
The TIES (Thermodynamic integration with enhanced sampling) protocol is a formally exact alchemical approach in computational chemistry [1], used for the rapid, accurate and reliable calculation of protein-ligand relative binding affinities. TIES employs ensemble simulations to capture and control the inevitable aleatoric and/or parametric uncertainties in such binding affinity calculations that makes it unique [2]. Now, for the first time, TIES is being released for open use.
TIES20, an updated ligand matching protocol with flexible topology superimposition algorithm employed using an exhaustive joint-traversal for identifying the maximum common sub-structure between ligand pairs, has been described and validated using the same dataset as the original TIES study. The major advancement here is the allowance of partial ring matching that reduces the size of the alchemical region on average resulting in significant improvement in the precision of results.
Funders:


![]()
![]()